This is an archival copy of the Visualization Group's web page 1998 to 2017. For current information, please vist our group's new web page.
High-throughput Characterization of Porous Materials
Problem Statement and Goals
Porous materials, such as zeolites and metal organic frameworks, are exploited in many current technologies and are considered to be a very important class of materials for many new industrial applications. Some of these new industrial applications include cracking catalysts in oil refinement, water softeners, and membranes or adsorbents for separations including carbon dioxide (CO2) capture applications. A key factor that determines the utility of any nanoporous material is its optimal pore topology along with the chemical composition for given conditions in a particular application. In an attempt to identify optimal materials for various applications, such as gas separations, researchers have started to screen large databases of porous materials. Molecular simulation techniques, such as the grand canonical Monte Carlo (GCMC) method, are often used in numerical simulations to accurately predict properties of materials and their guest-adsorption characteristics expressed as an experimentally verified adsorption isotherm. However, the computational cost of molecular simulations is high, significantly limiting the number of structures that can be analyzed. Furthermore, an accurate calculation of material properties depends on the correct classification of the accessibility of the pores in a material. Classification of the pore structure of materials typically involves visual inspection and becomes impractical with a large number of structures.Implementation and Results
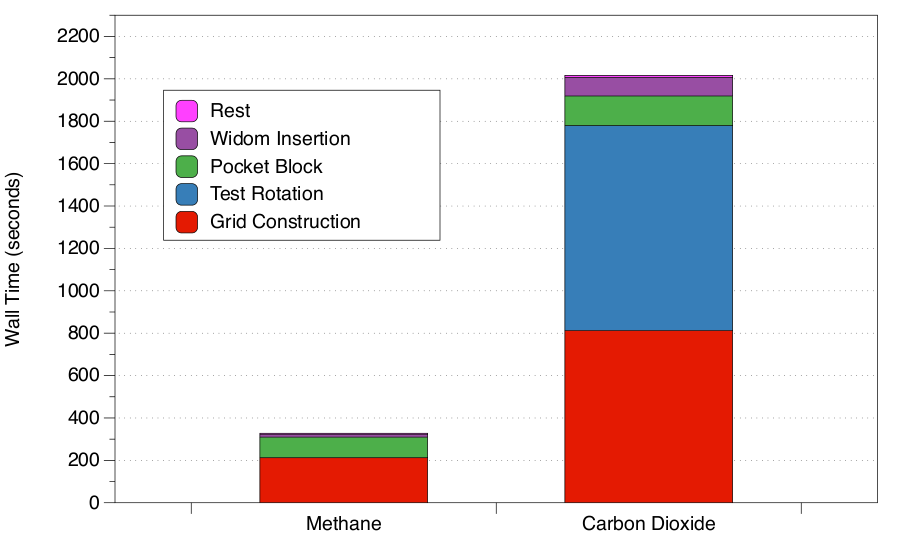
The goal of this work is to enable high-throughput screening of large material databases to characterize material properties and enable fast identification of candidate materials for application-specific problems such as carbon capture. As a means to characterize the zeolite structures, we compute the Henry coefficient (KH) and the heats of adsorption ( Δhi) values for carbon dioxide (CO2) and methane (CH4). These quantities characterize adsorption of the gas molecules in porous materials. KH is a basic constant that relates the equilibrium between the gas and the adsorbed phase, and hence describes adsorption at a very low pressure regime. In the context of CO2 capture, an ideal material would exhibit a large CO2 KH compared to the KH values of other flue gases, resulting in high selectivity for CO2.High-performance Material Simulations: In order to process large sets of materials within a reasonable time, our molecular simulation tool utilizes graphical processing units (GPUs) to efficiently conduct parallel calculations. In order to be able to efficiently utilize multiple GPUs in distributed computing environments, we developed a MPI-based task scheduler to assign and balance computing tasks among multiple compute nodes---with each task consisting in the evaluation of single molecular structures. Figure 1 summarizes the performance of our algorithm for computing the KH and Δhi for 144 experimentally verified zeolite structures from the IZA database.
Pocket Blocking: In practice, materials often contain inaccessible pockets, i.e., regions of space a guest molecule could occupy, but which cannot be accessed by guest molecules due to the positions of the surrounding atoms. In computer calculations, it is critical to account for such inaccessible pockets to avoid false biases in the calculation of guest-accessible volumes/surface areas, or the prediction of adsorption properties using molecular simulation techniques. To enable accurate, high-throughput characterization for large numbers of materials, we have developed automated methods for the segmentation and classification of the void space of molecules into accessible and inaccessible regions.
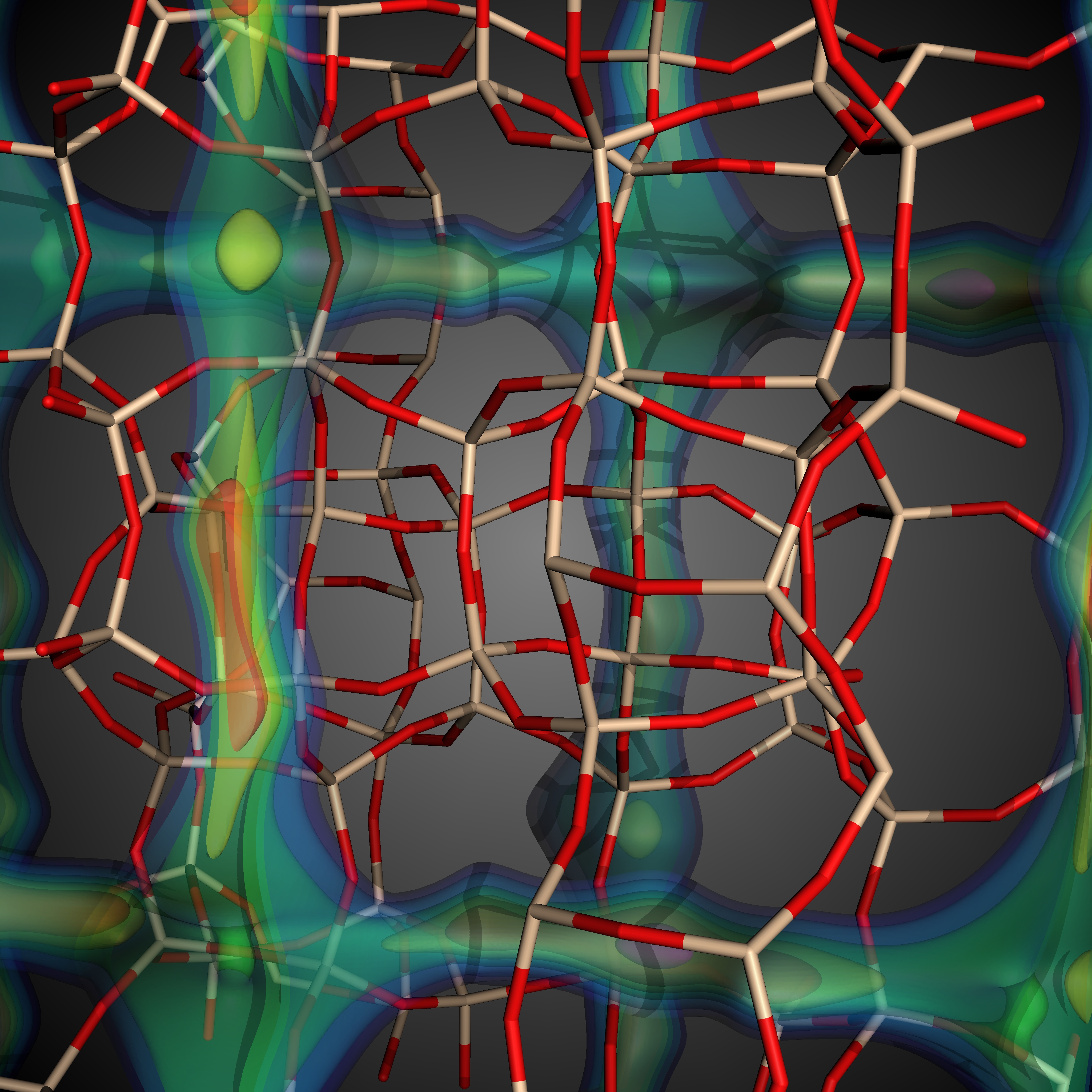
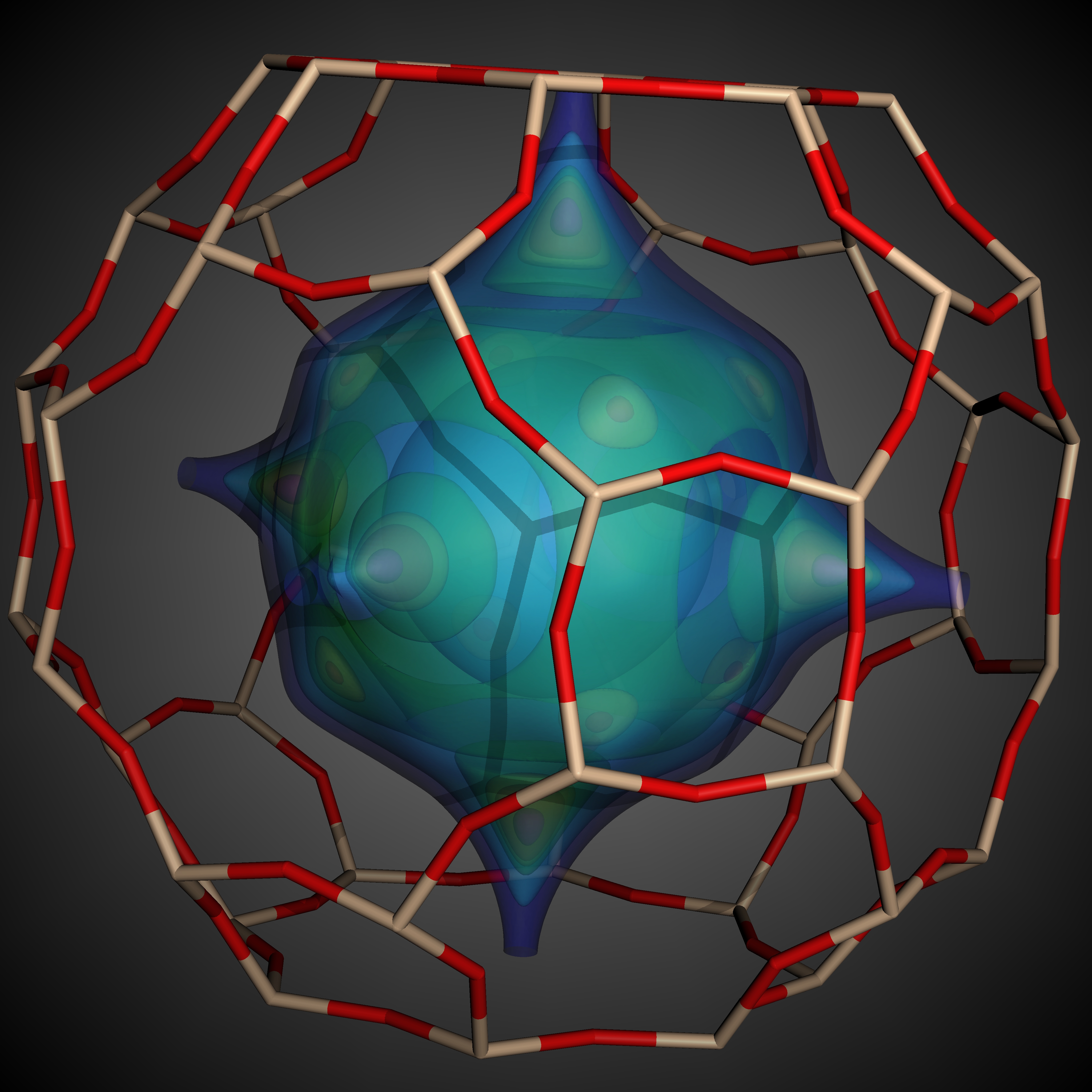
Visualization: Aside from computing Henry coefficients (KH) and heats of adsorption (Δhi), we can further analyze individual structures in the simulations by visualizing the local Henry coefficient values---to determine the local adsorption property of a given material---and the energy grid, describing at each discrete grid location the total Lennard-Jones and Coulomb potentials between the gas molecule and all of the framework atoms that make relevant contributions to the interaction. We use the state-of-the-art, high-performance, parallel visualization system VisIt for this purpose (see Figure 3).
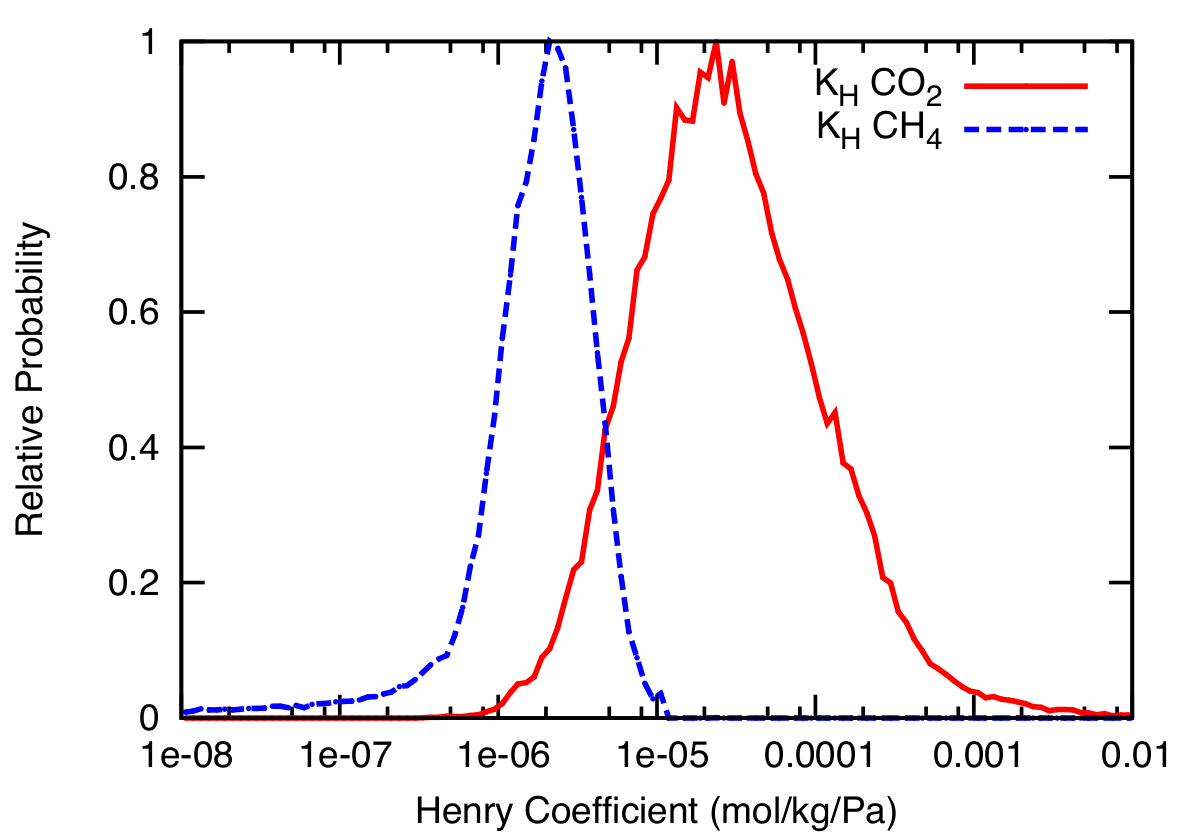
Characterization of Zeolites: Using the approach described here, we processed 135,224 hypothetical zeolites in the database. Figure 2 shows the histogram of both the CO2 and CH4 Henry coefficient (KH) values for all of the zeolite structures. We observe that the KH values for CO2 are in general higher than that of CH4---further confirming why zeolites are seen as one of the ideal candidates for carbon capture. The broader distributions of the CO2 KH values also indicates that the range of possible structures with different CO2 adsorption properties remains large compared to CH4. Extrapolating from detailed simulation times obtained from the IZA structures (see Figure 1), we can obtain the CO2 KH values for the entire hypothetical zeolite structures in about 50 hours of total wall time, utilizing 8 Tesla C2050 GPUs of the NERSC GPU cluster Dirac.
This effort on the simulation of porous materials has been led by Jihan Kim, Richard L. Martin, Maciej Haranczyk, and Berend Smit. This effort has been supported by Oliver Rübel, of the Visualization Group, in particular, with respect to development of distributed computing using MPI and visualization. This work is presented in more detail in a joint 2012 JCTC journal paper by Jihan Kim et al. [1].
 |
 |
| Figure 1: Timing results for calculation of the Henry coefficient (KH) and heats of adsorption (Δhi) for CH4 and the CO2
for the 188 experimentally verified IZA zeolite structures. The total
computational time is divided by the major routines that comprise the
simulation code. As these results show, using our high-performance
molecular simulation tool enables screeningof large numbers of molecular structures in a short amount of time. ( Image courtesy of Kim et al. [1]) |
Figure 2: Distribution of CO2 and CH4 Henry coefficient (KH) values for 135,224 hypothetical zeolite structures. In general, the CO2 KH values are larger than the CH4 KH, indicating that zeolites have high selectivity for CO2, confirming why zeolites are seen as one of the ideal candidates for carbon capture. (Image courtesy of Kim et al. [1]) |
 |
 |
| (a) MFI | (b) LFA |
| Figure
3: Visualization of MFI and LTA showing i) the molecular bonding
structure, and ii) isosurface visualization of the energy landscape.
Low energy values (warmer colors) indicate that less energy is needed
to insert the donor molecule (here methane) at the given location. (Image generated by Richard L. Martin using VisIt) |
|
Impact
By approaching the problem of screening large numbers of materials from a high-performance computing view and by utilizing fast molecular simulation techniques, we have been able to characterize a very large set of porous materials---more than an order of magnitude larger than what has been reported previously. Additionally, we have addressed the problem of characterizing the inner pore structure (and the accessibility of pores) for large sets of materials.Our code has the capability to quickly compute the Henry coefficient and the heats of adsorption values for many different gas molecules immersed inside a porous material. We can further analyze individual structures in the simulations by visualizing local Henry coefficient values to determine local adsorption properties of a given material. Although the simulation results presented in this work pertain to zeolites, the code can be easily extended to process other classes of microporous materials. The predicted molecular properties obtained from the simulation code can provide valuable insights for computational chemists and experimentalists to identify structures of interest for further computational analyses and for synthesizing materials inside a large porous materials database.